
~Research interests~
1. Engineering and design of antimicrobial peptides.
(抗菌胜肽研發與應用)
2. Structure Guided Discovery of PD-1PD-L1 Interaction Inhibitors Peptide Design, Screening, and
Optimization via Computation-Aided Phage Display Engineering.
(以噬菌體工程與人工智慧蛋白分析開發抗PD1/PDL1結合之阻斷劑用於癌症之細胞療法)
3. Using phage-display protein library for screening specific protein as novel CD73 inhibitors and
application to cancer therapy.
(利用噬菌體蛋白庫篩選特異性蛋白作為新型CD73抑制劑應用於癌症治療)
4. Comprehensively Developing a phage display platform for screening antibody-like scaffold
protein library targeting ACVR2A/ACVR2B to control and relieve Obesity.
(噬菌體工程建構類抗體蛋白變異庫來開發靶向 ACVR2A/2B 之蛋白藥-應用於控制與緩解肥胖)
5. Development of highly potent NP/TCM-based inhibitor against human glutaminyl cyclase for
Alzheimer's disease treatment.
(開發人類麩酸胺環化酶天然物與中藥抑制劑應用於阿茲海默症之治療)
6. Pharmacophore-Based Inhibitor Screening Targeting human Lp-PLA2 for Diabetic Macular Edema and Alzheimer's
Disease Treatments.
(開發人類脂蛋白相關蛋白酶A之抑制劑應用於糖尿病眼底病變與阿茲海默症之治療)
7. Drug development of transnational medicine for tjhe treatments of cancer and diabetes.
(轉譯醫學藥物研發~治療癌症與糖尿病)
噬菌體胜肽蛋白工程開發新藥

實驗室學習
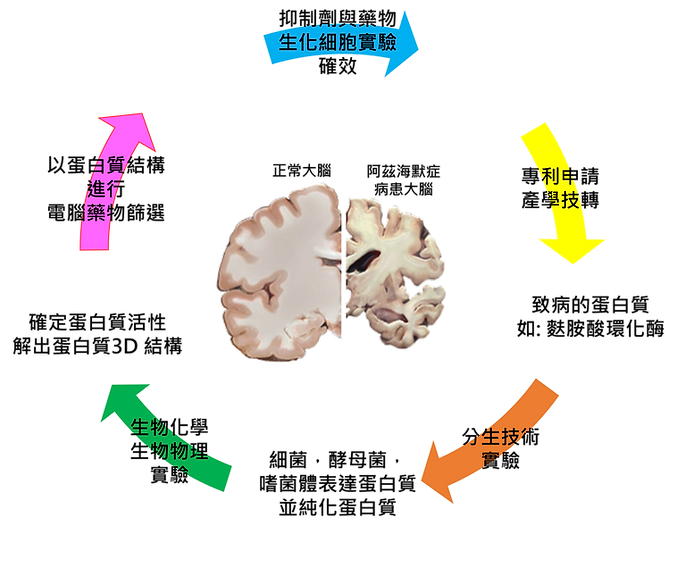
以上圖為例子,我們實驗室主要研究目的為新藥開發。目前研究的疾病有阿茲海默症`,抗癌藥,以及抗菌藥物。以阿茲海默症為例子,我們找出導致阿茲海默症的致病蛋白質後,會進行以下實驗 (也就是你進來能學到的技術)。
分生技術
利用分生技術,把致病蛋白基因轉置於細菌能利用的載體上,並轉型到細菌體內,讓細菌能幫我們表達出這個致病蛋白讓我們來研究。後續也利用蛋白質純化技術來純化高純度蛋白。
生物化學,生物物理實驗
有了蛋白質之後,我們首先確定蛋白質的活性,利用到生化實驗技術。
確定蛋白的活性後,接著用運生物物理實驗來把蛋白質的結構 (也就是它的長相,形狀,輪廓) 解析出來。
慢著慢著~還沒結束!!!!!! 我們解出這個致病蛋白質的結構,不是為了看一看玩一玩而已。我們要運用這個蛋白質結構來進行 AI 電腦藥物設計開發 !!!!!
AI 電腦藥物篩選
我們把蛋白質的輪廓,外型,以及它的樣子輸入給電腦,電腦 AI 就會以這個蛋白質的身體 (三度空間的結構),來找出能夠跟這的蛋白質結合的小分子抑制劑 (藥物)。
生化細胞實驗確效
電腦協助你篩出藥物後,接下來重要的階段就是驗證這個篩出來的小分子是不是能真的抑制蛋白質的
活性 !!!!! 我們利用到生化實驗以及細胞實驗來確認效果 !!!!!
後續動物實驗也將委託有國家級認證的動物研實驗中心究來協助我們確認藥效。
專利申請與產學技轉 (由老師執行)
如果你研究的抑制劑很有效,就可以申請專利保護。並且可以技術轉移給廠商,以及進行產學合作 !!!!
(這部分老師也是很有經驗的 !!!!)。此外,最重要的是,你的論文以及畢業都不是問題 ^^
~以上就是,你進來實驗室能學到的實驗技術內容~
Video introduction of student research results
學生研究成果之影片介紹
(1)
(2)
(3)
(4)
(5)
研究成果發表~許心冕同學
題目: 開發有潛力的人類甘醯基甘油脂肪酶抑制劑來治療或緩解神經炎症
Title: Development of Potential Human Monoacylglycerol Lipase Inhibitors for the Treatment or Alleviation of Neuroinflammation.
研究成果發表~張亦璿同學
題目: 開發新型麩胺酸環化酶抑制劑來治療阿茲海默症
Title: Development of Novel Glutaminyl Cyclase Inhibitors for the Treatment of Alzheimer's Disease.
研究成果發表~林子嵂同學
題目: 抗菌胜肽抑菌機制之探討
Title: Investigation of the Antimicrobial Mechanism of Action of Antimicrobial Peptides.
近期研究內容成果 (尚未更新,老師 (身兼網頁工程師) 太忙了~
詳見我們的研究成果 paper 發表~ ^^ ~
COVID-19 Spike protein 突變熱點研究
Comprehensive Deep Mutational Scanning Reveals the Immune-Escaping Hotspots of SARS-CoV-2 Receptor-Binding Domain Targeting Neutralizing Antibodies
The rapid spread of SARS-CoV-2 has caused the COVID-19 pandemic, resulting in the collapse of medical care systems and economic depression worldwide. To combat COVID-19, neutralizing antibodies have been investigated and developed. However, the evolutions (mutations) of the receptor-binding domain (RBD) of SARS-CoV-2 enable escape from neutralization by these antibodies, further impairing recognition by the human immune system. Thus, it is critical to investigate and predict the putative mutations of RBD that escape neutralizing immune responses. Here, we employed computational analyses to comprehensively investigate the mutational effects of RBD on binding to neutralizing antibodies and angiotensin-converting enzyme 2 (ACE2) and demonstrated that the RBD residues K417, L452, L455, F456, E484, G485, F486, F490, Q493, and S494 were consistent with clinically emerging variants or experimental observations of attenuated neutralizations. We also revealed common hotspots, Y449, L455, and Y489, that exerted comparable destabilizing effects on binding to both ACE2 and neutralizing antibodies. Our results provide valuable information on the putative effects of RBD variants on interactions with neutralizing antibodies. These findings provide insights into possible evolutionary hotspots that can escape recognition by these antibodies. In addition, our study results will benefit the development and design of vaccines and antibodies to combat the newly emerging variants of SARS-CoV-2.
近年來新冠肺炎肆虐全球,對全世界人類的健康造成重大威脅。如今雖然已經有對抗新冠肺炎的疫苗以及口服用藥,但此病毒不換的演化突變,降低了這些預防以及治療之效果。為了要提前預知此新冠病毒的突變路徑與結果,本研究利用結構生物學以及計算生物的方法來探討新冠肺炎病毒蛋白 Spike protein 其受體結合蛋白 (receptor binding domain) 的突變熱點。Spike 蛋白是新冠肺炎病毒在感染人體時主要會接觸人類受體 (ACE2) 的主要感染媒介。Spike 蛋白的突變導致疫苗的失效。我們的研究利用目前已經被解出的 Spike蛋白與抑制他的抗體結構複合體來進行生物計算分析。我們將 Spike 蛋白的關鍵胺基酸 (會與抗體作用的胺基酸) 進行突變,將這些胺基酸突變成其他 20 種胺基酸,並進行 Spike 蛋白與抗體親和力之分析。結果得知 Spike 蛋白的胺基酸 K417, L452, L455, F456, E484, G485, F486, F490, Q493, S494 如果突變成其他種胺基酸就會破壞此蛋白與抗體的親和性,導致抗體沒有作用,病毒因此躲避免疫系統並繼續存活。此研究成果,揭露 Spike 蛋白的可能突變熱點。我們的研究成果對後續對抗新冠肺炎之疫苗以及抗體的研究提供重要性與關鍵性的訊息!

FIGURE 1. The mutational binding stabilities of RBD variants interacting to H11-D4 nanobody. (A) The molecular interactions of SARS-CoV-2 RBD with H11-D4 nanobody analyzed by ligplot. The chains E and F correspond to RBD and H11-D4 nanobody, respectively. (B) The heatmap of interactive residues of RBD derived from the calculated mutational binding stabilities by using Discovery Studio 3.5 (DS), Mutabind2, FoldX, and mCSM-PPI2. The boxes of each mutations were colored with the gradient of a range between blue (stabilized binding) and red (destabilized binding). In all panels, the solid and hollow circles denote significant and moderate decreases of the binding stabilities, respectively.
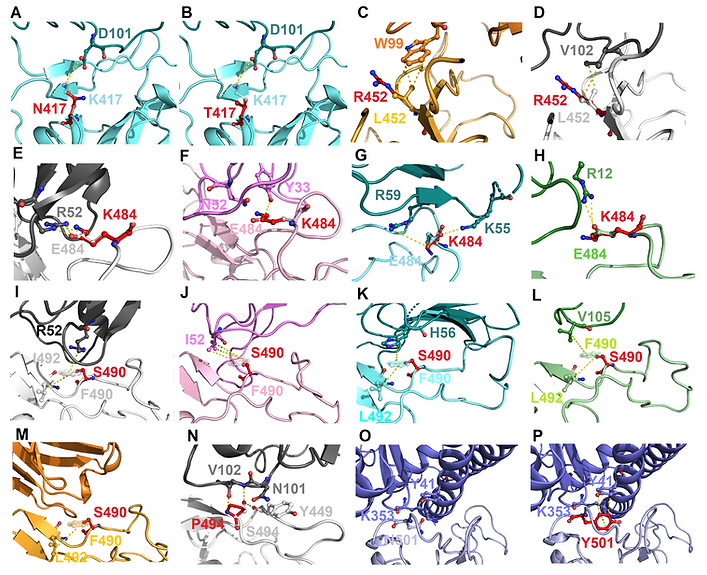
FIGURE 2. The molecular interactions of SARS-CoV-2 RBD variants identified from pandemic isolates. (A) The compared intermolecular interactions of K417 (light cyan stick) and its variant N417 (red stick) in the complex of RBD–MR17. (B) The overlapped view of intermolecular interactions of K417 (light cyan stick) and its variant N417 (red stick) in the complex of RBD–MR17. (C) The compared intermolecular interactions of L452 (light orange stick) and its variant R452 (red stick) in the complex of RBD–SR4. (D) The overlapped view of intermolecular interactions of L452 (light gray stick) and its variant R452 (red stick) in the complex of RBD–H11-D4. (E) The compared intermolecular interactions of E484 (light gray stick) and its variant K484 (red stick) in the complex of RBD–H11-D4. (F) The overlapped view of intermolecular interactions of E484 (light pink stick) and its variant K484 (red stick) in the complex of RBD–VH1-2-15. (G) The intermolecular interactions of E484 (light cyan stick) and its variant K484 (red stick) in the complex of RBD–MR17. (H) The intermolecular interactions of E484 (light green stick) and its variant K484 (red stick) in the complex of RBD–P2B-2F6. (I) The compared intermolecular interactions of F490 (light gray stick) and its variant S490 (red stick) in the complex of H11–D4. (J) The overlapped view of intermolecular interactions of F490 (light pink stick) and its variant S490 (red stick) in the complex of RBD–VH1-2-15. (K) The intermolecular interactions of F490 (light cyan stick) and its variant S490 (red stick) in the complex of RBD–MR17. (L) The intermolecular interactions of F490 (light green stick) and its variant S490 (red stick) in the complex of RBD–P2B-2F6. (M) The intermolecular interactions of F490 (light orange stick) and its variant S490 (red stick) in the complex of RBD–SR4. (N) The intermolecular interactions of S494 (light gray stick) and its variant P494 (red stick) in the complex of RBD–H11-D4. (O) The intermolecular interactions of N501 (light purple stick) in the complex of RBD–ACE2. (P) The intermolecular interactions of Y501 (red stick) in the complex of RBD–ACE2. In all panels, proteins are shown in ribbons and the molecular interactions are presented as dash lines.
抗菌胜肽設計與結構功能解析
Engineering and design of antimicrobial peptides
Background and highlights:
The growing emergence of antibiotic-resistant microbial strains, putting considerable threats to public health, makes an urging need to develop new anti-infective molecules. Natural antimicrobial peptides (AMPs), regarded as a novel class of antibiotics, failed in clinical trials due to the drawbacks in hemolytic activity, cytotoxicity, and degradation. Nowadays, rationally designed and synthetic AMPs are believed to overcome the limitations of native peptides. A synthetic peptide GW-Q6 with enhanced antimicrobial activity, decreased hemolytic activity, and improved selectivity, draws our attention to investigate its mode of action. Here, we employed biochemical and biophysical techniques together with MD simulation to explore the structure-function relationship of GW-Q6. Far-UV CD and NMR studies demonstrated GW-Q6 is a well-defined a-helical structure in DPC micelles. We determined the first solution structure of GW-Q6 which is of amphipathic nature in DPC micelles. The paramagnetic relaxation enhancement (PRE) investigation disclosed that GW-Q6 orients its a-helix segment (K7-K17) buried into DPC micelles. The calcein leakage assay uncovered that GW-Q6 is highly selective against negatively charged bacterial membranes. Additionally, the a-helix segment (K7-K17) is critical for membrane-permeabilization and antimicrobial activity. Moreover, the conserved lysine residues K3, K7, and K14 could be critical for helical formation and membrane binding affinity. The aromatic residues Y19 and W20 are functionally important to position the C-terminus on the surface of the micelle. Taken together, our study provides insights into the mode of action and residue functional annotation of GW-Q6, which is of great helpful in modifying and developing potent AMPs as the therapeutic used antibiotics.
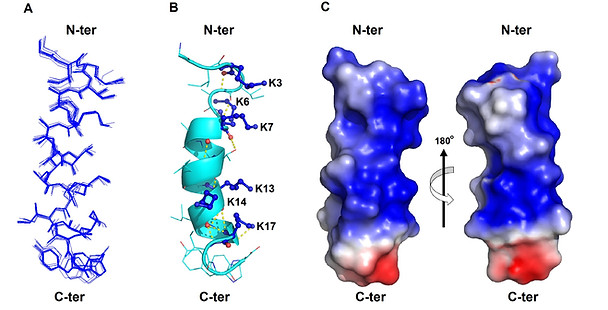
Figure 1. The solution structure of GW-Q6 determined in DPC micelles.
近年來由於抗生素的濫用,導致了許多抗藥性病原菌快速崛起。多重抗藥性細菌株引發高致病與致死率,已對全球人類的健康造成極大威脅。因此,開發新穎的抗菌藥物,刻不容緩。然而天然的抗菌胜肽具有顯著的殺菌效果,而且可借由循理性的胜肽設計來增強其抗菌能力。因此,有殺菌效果的抗菌胜肽已被視為極具有潛力的新型抗生素研發標的。抗菌胜肽擁有多重抗菌機制,不易使微生物產生抗藥性。因此,適合用於抗藥性微生物感染疾病的治療。本計畫目的在於研究合成性抗菌胜肽 (synthetic antimicrobial peptide) 其殺菌功能與機制。在此計畫研究中,將以生物化學,生物物理學與結構生物學的角度,分析並探討抗菌胜肽的結構與其抗菌機制間的相關性並根據所得結果來優化抗菌胜肽的殺菌效果。我們將試驗三種抗菌胜肽與脂質體 (liposome) 及脂肪微胞 (micelle) 的交互作用,並以圓二色偏極光譜 (circular dichroism) 分析法來觀測胜肽二級結構生成的現象與程度。同時也測定此三種抗菌胜肽對不同帶電性質脂質體的通透性與選擇性,來了解合成性抗菌胜肽其辨識真核細胞膜及細菌細胞膜的能力。此外,我們也會進行革蘭氏陽性菌及革蘭氏陰性菌細胞膜通透性分析,藉此比較三種抗菌胜肽的破膜殺菌效果。根據上述實驗的研究結果,選出破壞細菌細胞膜能力佳並且具有選擇性的抗菌胜肽進行結構分析。研究將透過核磁共振光譜 (NMR spectroscopy) 分析抗菌胜肽鑲嵌於脂肪微胞上的 3D 立體結構。最後在完成結構分析後,對於抗菌胜肽序列上有作用於脂肪微胞的功能性胺基酸序列進行重組,再以脂肪微胞通透性分析試驗重組胜肽之活性。
New antibacterial is urgently needed. We conducted a protein-DNA complex-guided pharmacophore approach to screen inhibitors against the response regulator, PmrA, of polymyxin B-resistant Klebsiella pneumoniae (KP). The identified lead, E1 (IC50 = 10.2 µM), targets the DNA binding domain of PmrA (KD = 1.7 µM), where the conserved residues, R171, R198, K203, and Y214 are hotspots for antimicrobial development. Treatment of E1 restored the susceptibility of KP to polymyxin B.
克雷伯氏肺炎菌是常見的醫院內感染細菌,可引起肺炎、敗血症、腦膜炎、肝膿瘍、泌尿系統感染及腎膿瘍等,若治療不當則死亡率。此細菌目前衍生出多重抗藥性菌種,使得在治療上造成極大的困難。而 PmrA/PmrB 雙分子反應調控蛋白能調節克雷伯氏肺炎菌的抗藥性基因,使其對抗生素 polymyxin B 產生抗性。本研究以反應調控蛋白 PmrA-DNA的複合體結構為基礎,建構出藥效基團模型 (pharmacopoeia model)來篩選小分子抑制劑。研究結果發現,藥效基團模型Ch2pharN4 能篩選到一個強效的抑制劑分子 E1 (IC50 = 10.2 uM)。此抑制劑 (E1) 會結合在 PmrA 的 DNA binding domain,進而阻止 PmrA 與標的基因的 promoter DNA 結合,進而削弱克雷伯氏肺炎菌對抗生素 (polymyxin B) 的抗藥性能力。此研究成果對未來研發新型抗細菌用藥提供重要性的訊息。
藥物設計開發
Protein DNA Complex‐Guided Discovery of an Antibacterial Lead E1 in Restoring the Susceptibility of Klebsiella Pneumoniae to Polymyxin B by Targeting Response Regulator PmrA
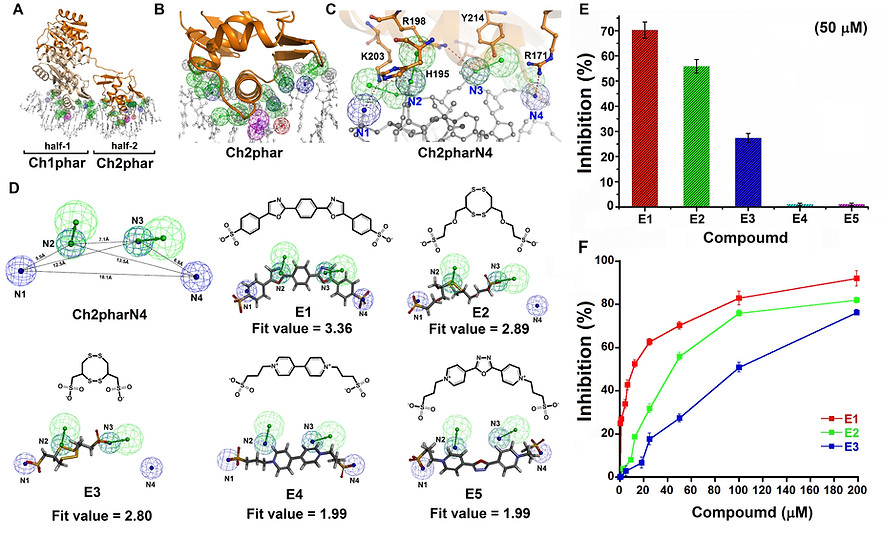
Figure 1. Pharmacophore-based inhibitor screening and functional examination. (A) Receptor-ligand pharmacophore generation based on PmrA-DNA complex structure (PDB ID: 4S04) and the generated pharmacophore feature clusters, Ch1phar and Ch2phar. (B) The amplified view of Ch2phar. (C) The Ch2pharN4 pharmacophore scaffold contains four negative charged groups (N1-N4). The complimentarily interactive residues (orange sticks) of PmrA toward Ch2pharN4 are labelled. (D) The ligand pharmacophore mapping results of E1-E5. (E) The FP assay results with the additions of E1-E5 (50 µM). (F) Dose-dependent inhibitions of E1-E3 against PmrA-DNA complex formation determined by FP. (In panels A-D, the mesh sphere in red, blue, magenta, green, and dark-gray represent positive-charged, negative-charged, hydrogen-bond donor, hydrogen-bond acceptor, and excluded volume features, respectively. The PmrA are colored in wheat and orange; DNA are colored in white)

Figure 2. Determinations of binding site and dissociation constant of E1 toward PmrA. The overlapped 2D 1H-15N HSQC-TROSY spectra of (A) inactivated PmrA (black) titrated with E1 (green) and (B) activated PmrA (black) with E1 addition (red). (PmrA: E1 = 1:3 molar ratio; the purebred residues are labeled in black) (C) The perturbed residues of activated PmrA upon E1 binding were mapped onto the structure (cyan). (D) The overlapped 2D 1H-15N HSQC-TROSY spectra of PmrAC titrated with E1. The perturbed residues are labeled in black. (E) The amplified view of panel D. (F) The titration curves of backbone amide resonances of S167, V169, R207, V209, R210, G213, and Y214 were fitted to the equation valid for a 1 : 1 complex and the KD = 126 ± 34.6 µM. (G) The ITC binding isotherm for the interactions of E1 to PmrAC. (H) The BLI data showing the association and dissociation sensorgrams at distinct E1 concentrations for the interaction of PmrAC.

Figure 3. The complex structure of PmrA-E1 and the microbial viability assay results. (A) E1 binds in between a8-helix and C-terminal b-hairpin of DBD and is aligned with the pharmacophore scaffold, Ch2pharN4. (B) The residues of PmrA interacting with E1 are shown in sticks (magenta). (C) The 2D illustration of detailed interactions of E1 with PmrA. (D) The effects of E1 on the polymyxin B dose-dependent inhibition against KP.
此研究已申請中華民國專利,美國專利
